In een tijdperk waarin technologie de gezondheidszorg snel verandert en medische gegevens een steeds grotere rol spelen, is het begrijpen van hoe deze gegevens worden gebruikt, opgeslagen en beheerd, van cruciaal belang voor patiënten. Wij hebben een overzicht gemaakt van de meest voorkomende vragen die mensen hebben rondom hun medische gegevens en hun opslag.

- Waar worden mijn medische gegevens opgeslagen?
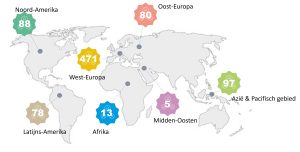
Medische gegevens kunnen op verschillende plaatsen worden opgeslagen, afhankelijk van waar en door wie ze worden verzameld. Dit kan variëren van elektronische patiëntendossiers in ziekenhuizen en huisartsenpraktijken tot speciale databanken en biobanken (waar lichaamsmateriaal wordt bewaard) voor onderzoeksdoeleinden.
Meer uitleg over de plekken waar medische gegeven worden opgeslagen vind je in het artikel ‘Waar wordt data opgeslagen’
- Hoe veilig zijn mijn medische gegevens?
Veiligheid en privacy zijn van het grootste belang als het gaat om medische gegevens. Daarom zijn zorginstellingen en onderzoeksorganisaties verplicht om strenge maatregelen te treffen om de vertrouwelijkheid en integriteit van deze gegevens te waarborgen. Zo moeten alle gegevens versleuteld worden opgeslagen, zijn er een strenge toegangscontroles en controleert men nauwkeurig op de naleving van wettelijke voorschriften zoals de AVG (Algemene Verordening Gegevensbescherming)
- Wie heeft toegang tot mijn medische gegevens?
Alleen geautoriseerde zorgverleners en onderzoekers hebben toegang tot medische gegevens. Denk hierbij aan artsen, verpleegkundigen, of medisch onderzoekers. Patiënten hebben doorgaans ook het recht om hun eigen medische gegevens in te zien en te beheren.
- Kan ik mijn medische gegevens zelf beheren?
Ja, steeds meer gezondheidszorgsystemen bieden patiënten de mogelijkheid om hun eigen medische gegevens te beheren via een zogenaamde Persoonlijke Gezondheidsomgeving (PGO). Zo’n platform stelt patiënten in staat om hun gezondheidsgegevens in te zien, te beheren en te delen met zorgverleners naar keuze.
- Wat gebeurt er met mijn medische gegevens na mijn behandeling?
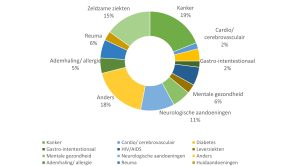
Na uw behandeling kunnen uw medische gegevens worden bewaard in het elektronisch patiëntendossier van de zorginstelling. Ze kunnen ook worden gebruikt voor wetenschappelijk onderzoek, alleen als je hiervoor toestemming hebt gegeven. In sommige gevallen kunnen gegevens geanonimiseerd worden en worden gebruikt voor epidemiologische studies of kwaliteitsverbeteringsinitiatieven.
- Hoe kan ik mijn toestemming voor het delen van mijn medische gegevens regelen?
Het is belangrijk om te weten dat je als patiënt controle hebt over wie toegang heeft tot jouw medische gegevens en voor welke doeleinden ze kunnen worden gebruikt. Je kan je toestemming voor gegevensdeling regelen door gebruik te maken van platforms zoals www.volgjezorg.nl of door contact op te nemen met je zorgverlener.
Het is belangrijk om je rechten en opties met betrekking tot je medische gegevens te begrijpen, zodat je goede beslissingen kunt nemen over je gezondheid. Als je nog vragen hebt over dit onderwerp, aarzel dan niet om contact op te nemen met je zorgverlener voor meer informatie. Jouw stem als patiënt is belangrijk en moet worden gehoord als het gaat om je gezondheid!